Quite often the onset of cold weather is accompanied by a weakening of the immune system. This in turn leads to a decrease in the body’s susceptibility to pathogens and the development of viral infections. Usually all diseases begin with a simple runny nose, so this symptom should not be ignored. Treatment of a cold consists of fighting the cause, which is caused by pathogens localized in the nasopharynx. Therefore, nasal antiviral agents are used to treat such conditions.
What is Oxolinic ointment?
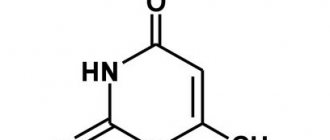
This is a pasty viscous substance based on oxoline. The ointment has a yellow tint, a homogeneous, slightly oily structure. Plastic, easily distributed over surfaces. After long-term storage, the product sometimes becomes pinkish or bluish. This feature does not affect its properties.
The medicine is released in different concentrations:
- with oxoline content 0.25%: in tubes of 10 g, used for the treatment of mucous membranes;
- with an active ingredient content of 3%: packaged in tubes of 10–30 g, suitable for treating skin or eliminating intense pathological processes.
Auxiliary compounds of the drug: wax, paraffin, vaseline oil.
How does Oxolinic ointment work?
The drug disrupts the integrity of the cell membranes of pathogens, causing their death. When applied to the surface of the skin and mucous membranes, the ointment mechanically prevents their infection, softens, and reduces the symptoms of inflammation. Oxolin affects pathogens of influenza, molluscum contagiosum, herpes simplex, adenoviruses, and rhinoviruses.
The use of ointment does not lead to a systemic effect on the body. The amount of oxoline that penetrates through the capillaries into the blood is insignificant. The product does not accumulate in tissues and has no cumulative effect. Particles that get inside are completely metabolized.
Types and principle of action of antiviral drugs
There are several forms of drugs to fight viruses. These are drops, spray and ointment. The most effective and common are drops based on human interferon. This drug has pronounced antiviral and immunomodulatory properties. Another active component is interferon inducers.
Antiviral drugs affect all stages of the introduction of pathogens. Some drugs help destroy them, while other drugs stop the production and spread of viruses in the body.
When to use Oxolinic ointment
Traditionally, the medicine is used during respiratory epidemics and when communicating with possible carriers of influenza viruses. The list of indications for use of the drug includes:
- influenza and other respiratory viral pathologies;
- blepharitis;
- inflammation of the conjunctiva;
- lichen lesions;
- molluscum contagiosum;
- dermatitis;
- herpes stomatitis;
- skin warts of viral origin.
The use of medicine for preventive purposes and at an early stage of pathologies is more effective than for advanced infections. To achieve a pronounced therapeutic effect, this drug must be combined with other drugs.
Instructions for use
Antiviral agents for nasal use are not recommended for use more than 2 times a year. They can cause a decrease in immune status, especially in childhood.
Regular use of medications leads to a decrease in the body's own human interferon. And the latter, in turn, becomes less susceptible to pathogenic pathogens - bacteria and viruses.
Any antiviral drugs should not be combined with nasal vasoconstrictor drops. This can cause severe dryness of the nasal mucosa.
Antiviral medications are strongly discouraged from being used as the primary method of therapy. They should be included in complex treatment, often together with oral forms.
Before instilling the product into the nasal passages, it is important to warm the bottle with the solution in your hands. If you have a profuse runny nose and severe congestion, you must first blow your nose, or better yet, rinse the nasal cavity with saline solutions.
For greater effectiveness of antiviral drugs, it is advisable to start using them as early as possible, before the first characteristic signs of the disease appear. Since the body begins to synthesize interferons no earlier than 3 days after the development of the pathology.
Side effects of the ointment
When first applied, a slight burning sensation, increased runny nose, and irritation of the integument are likely to occur. With prolonged daily use, the nasal mucosa may feel dry. The risk of drying out is especially high during simultaneous treatment with vasoconstrictor drops. With proper use of the product, the discomfort gradually goes away on its own. If symptoms increase over time, the ointment should be discarded, as an allergic reaction is possible.
Oxolinic ointment does not affect the speed of psychomotor reactions and the activity of the central nervous system; it is allowed when driving vehicles and operating complex equipment.
Antiviral drugs in general practice
Antiviral drugs are compounds of natural or synthetic origin used to treat and prevent viral infections. The action of many of them is selectively aimed at various stages of the development of viral infection and the life cycle of viruses.
Currently, more than 500 viruses are known to cause human diseases. Viruses contain single- or double-stranded ribonucleic acid (RNA) or deoxyribonucleic acid (DNA) enclosed in a protein shell called a capsid. Some of them also have an outer shell of lipoproteins. Many viruses contain enzymes or genes that enable reproduction in the host cell. Unlike bacteria, viruses do not have their own metabolism: they use the metabolic pathways of the host cell.
RNA viruses either synthesize messenger RNA (mRNA), or the RNA itself performs the function of mRNA. It synthesizes viral proteins, including RNA polymerase, with the participation of which viral mRNA is formed. Transcription of the genome of some RNA viruses occurs in the nucleus of the host cell. Under the influence of retroviral reverse transcriptase, complementary DNA (provirus) is synthesized based on viral RNA, which is integrated into the genome of the host cell. Subsequently, during transcription, both cellular RNA and viral mRNA are formed, on which viral proteins are synthesized for the assembly of new viruses. Viruses and the diseases they cause are shown in Table. 1.
Basic mechanisms of action of antiviral drugs
At the infection stage, the virus is adsorbed on the cell membrane and penetrates the cell. During this period, drugs are used that disrupt this process: soluble false receptors, antibodies to membrane receptors, inhibitors of the fusion of the virus with the cell membrane.
At the stage of virus penetration, when the virion is deproteinized and the nucleoprotein is “undressed,” ion channel blockers and capsid stabilizers are effective.
At the next stage, intracellular synthesis of viral components begins. At this stage, inhibitors of viral DNA polymerases, RNA polymerases, reverse transcriptase, helicase, primase, and integrase are effective. The translation of viral proteins is affected by interferons (IFN), antisense oligonucleotides, ribozymes and inhibitors of regulatory proteins. Proteolytic cleavage is affected by protase inhibitors.
IFN and inhibitors of structural proteins actively affect virus assembly.
The final stage of the replication cycle involves the release of daughter virions from the cell and the death of the infected host cell. At this stage, neuraminidase inhibitors, antiviral antibodies and cytotoxic lymphocytes are effective.
There are different classifications of antiviral agents. This article presents a classification based on the effect on a particular virus (Table 2).
Let's look at anti-influenza and anti-herpetic drugs.
Classification of antiviral drugs approved for use in Russia.
- group of anti-influenza drugs: – Amantadine; – Arbidol; – Oseltamivir; - Rimantadine.
- Drugs acting on herpes viruses: – Alpizarin; – Acyclovir; – Bonafton; – Valacyclovir; – Ganciclovir; – Glycyrrhizic acid; – Idoxuridine; – Penciclovir; – Riodoxol; – Tebrofen; – Tromantadine; – Famciclovir; - Florenal.
- Antiretroviral drugs: – Abacavir; – Amprenavir; – Atazanavir; – Didanosine; – Zalcitabine; – Zidovudine; – Indinavir sulfate; – Lamivudine; – Nelfinavir; – Ritonavir; – Saquinavir; – Stavudin; – Phosphazide; - Efavirenz.
- Other antiviral drugs: – Inosine pranobex; – Interferon alpha; – Interferon alpha-2; – Interferon alpha-2b; – Interferon beta-1a; – Interferon beta-1b; – Yodantipyrine; – Ribavirin; – Tetraoxo-tetrahydronaphthalene (Oxolin); – Tiloron; – Flacoside.
Anti-influenza drugs (Table 2)
Arbidol is a derivative of indole carboxylic acid. The mechanism of action of the drug consists of suppressing the reproduction of the influenza virus, influencing the synthesis of IFN, increasing the number of T-lymphocytes and the functional activity of macrophages, as well as an antioxidant effect.
The drug penetrates unchanged into both uninfected and infected cells and is detected in the nuclear and cytoplasmic fractions. Arbidol inhibits the process of fusion of the lipid viral envelope with endosome membranes (at pH 7.4), leading to the release of the viral genome and the beginning of transcription. Unlike amantadine and rimantadine, Arbidol inhibits the release of the nucleocapsid itself from external proteins, neuraminidase and the lipid membrane. Thus, Arbidol acts in the early stages of viral reproduction.
The drug has no strain specificity (in cell cultures it suppresses the reproduction of the influenza A virus by 80%, the influenza B virus by 60% and the influenza C virus by 20%, and also affects the avian influenza virus, but weaker than the reproduction human strains of influenza virus).
IFN synthesis increases, starting from taking 1 tablet to 3 tablets. However, there is no further increase in IFN levels when taking Arbidol. A rapid increase in IFN synthesis can have a preventive effect when taken before the onset of influenza.
Arbidol has an immunomodulatory effect, leading to an increase in the total number of T-lymphocytes and T-helper cells. Moreover, normalization of these indicators was observed in patients with an initially reduced number of CD3 and CD4 cells, and in individuals with normal functioning of the cellular component of immunity there were practically no changes in the number of T-lymphocytes and T-helper cells. At the same time, the use of Arbidol does not lead to a significant decrease in the absolute number of T-suppressor lymphocytes - thus, the stimulating activity of the drug is not associated with inhibition of the function of suppressor cells. Arbidol increases the total number of macrophages with engulfed bacteria and the phagocytic number. It is assumed that the activating stimuli for phagocytic cells were cytokines and, in particular, IFN, the production of which is enhanced under the influence of the drug. The content of natural killer cells, NK cells, also increases, which allows the drug to be characterized as an inducer of natural killer cell activity.
The drug is quickly absorbed from the gastrointestinal tract (GIT). T1/2 is 16–21 hours. It is excreted unchanged in feces (38.9%) and urine (0.12%). During the first day, 90% of the administered dose is eliminated.
Drug interactions of Arbidol with other drugs have not been described in the literature.
Almost the only side effects of the drug are allergic reactions. The drug is approved for use from 2 years of age.
Arbidol has a fairly wide spectrum of antiviral action and is used for the prevention and treatment of influenza types A and B, including those complicated by bronchitis and pneumonia; acute respiratory diseases (ARVI); chronic bronchitis, pneumonia, recurrent herpetic infection; in the postoperative period - to normalize the immune status and prevent complications.
Amantadine and rimantadine are adamantane derivatives. Both drugs suppress the reproduction of virus A, even in small doses. Their antiviral activity is due to two mechanisms.
Firstly, they act at the early stage of viral reproduction, suppressing the “undressing” of the virus. The primary target for these drugs is the M2 protein of the influenza A virus, which forms an ion channel in its envelope. Suppression of the function of this protein leads to the fact that protons from endosomes cannot enter the virus, the dissociation of the ribonucleide and the release of the virus into the cytoplasm are blocked.
Secondly, they can also act at the stage of virus assembly, apparently by changing the processing of hemagglutinin. This mechanism is possible in some strains of viruses.
Among wild strains, drug resistance rarely occurs, but resistant strains are obtained from patients taking them. The sensitivity and resistance of viruses to amantadine and rimantadine are cross-sensitivity.
Both drugs are well absorbed when taken orally and have a large volume of distribution. Most amantadine is excreted unchanged in the urine. The half-life (T1/2) in young people is 12–18 hours, in the elderly it almost doubles, and in renal failure it increases even more. Therefore, the dose of the drug must be reduced even if there is a slight change in renal function. Rimantadine is actively metabolized in the liver, T1/2 averages 24–36 hours, 60–90% of the drug is excreted in the urine in the form of metabolites.
When taking both drugs, minor dose-dependent disturbances in the gastrointestinal tract (nausea, loss of appetite) and central nervous system (CNS) (irritability, insomnia, impaired concentration) are most often noted. When taking high doses of amantadine, significant neurotoxic effects are possible: confusion, hallucinations, epileptic seizures, coma (these effects may be enhanced by concomitant use of H1-blockers, M-anticholinergics, psychotropic drugs and ethanol). Safety of use during pregnancy has not been established. Allowed for use from 7 years of age.
The drugs are used to prevent and treat influenza A. Taking them during influenza epidemics allows one to avoid infection in 70–90% of cases. In persons with uncomplicated influenza A, treatment with drugs for 5 days in age-specific dosages, started at an early stage of the disease, reduces the duration of fever and general symptoms by 1–2 days, accelerates recovery and sometimes shortens the period of virus shedding.
Oseltamivir is an inactive precursor that is converted in the body to an active metabolite, oseltamivir carboxylate. It is a transition analogue of sialic acid and a selective inhibitor of neuraminidase of influenza A and B viruses. In addition, it suppresses strains of influenza A virus that are resistant to drugs derived from adamantane.
Neuraminidase of the influenza virus cleaves off the terminal residues of sialic acids and, thus, destroys receptors located on the surface of cells and new viruses, i.e., it promotes the exit of the virus from the cell upon completion of reproduction. The active metabolite of oseltamivir causes changes in the active site of neuraminidase and suppresses its activity. Viruses aggregate on the cell surface and their spread slows down.
Resistant strains of influenza A virus are found in 1–2% of patients taking the drug. To date, no resistant strains of influenza B virus have been detected.
When taken orally, the drug is well absorbed. Eating does not affect its bioavailability, but reduces the risk of side effects on the gastrointestinal tract. The drug undergoes enzymatic hydrolysis in the gastrointestinal tract and liver with the formation of an active metabolite. The volume of distribution of the drug approaches the volume of fluid in the body. T1/2 of oseltamivir and its active metabolite is 1–3 and 6–10 hours, respectively. Both compounds are excreted primarily by the kidneys unchanged.
When taken orally, minor abdominal discomfort and nausea are possible, which decrease when taking the drug with food. Gastrointestinal disorders usually resolve within 1–2 days, even if the patient continues to take the drug. No clinically significant interactions of oseltamivir with other drugs have been identified. The drug is used in children over 1 year of age.
Oseltamivir is used to treat and prevent influenza. Prophylactic administration of oseltamivir during epidemics reduces the incidence both among those vaccinated with influenza vaccine and among unvaccinated people. When treating influenza with this drug, recovery occurs 1–2 days earlier, and the number of bacterial complications is reduced by 40–50%.
Antiherpetic drugs
Before moving on to a discussion of antiherpetic drugs, it is necessary to recall the various herpes viruses and the diseases caused by them (Table 4). Unfortunately, the arsenal of modern antiviral drugs does not contain drugs that act on all herpes viruses simultaneously (Table 5).
Herpes simplex virus type 1 causes damage to the skin, mouth, esophagus and brain; herpes simplex virus type 2 causes damage to the external genitalia, rectum, skin and meninges. The first antiherpetic drug approved for use was vidarabine (1977). However, due to its high toxicity, it was used to treat diseases caused by the herpes simplex virus and Varicella-zostervirus, only for health reasons. Since 1982, acyclovir has been used to treat patients with less severe disease.
Acyclovir is an acyclic analogue of guanosine, and valacyclovir is the L-valine ester of acyclovir. Acyclovir inhibits viral DNA synthesis after phosphorylation by viral thymidine kinase within infected cells. The acyclovir triphosphate formed in the cell is integrated into the DNA chain synthesized in the host cell, which leads to the cessation of growth of the viral DNA chain. The DNA molecule, which contains acyclovir, binds to DNA polymerase, irreversibly inactivating it.
Viral resistance may result from decreased viral thymidine kinase activity and changes in viral DNA polymerase. Changes in enzyme activity occur as a result of mutations.
The bioavailability of acyclovir when taken orally is only 10–30% and decreases with increasing dose. Unlike acyclovir, the bioavailability of valacyclovir when taken orally reaches 70%. The drug is quickly and almost completely converted to acyclovir. Acyclovir penetrates into many biological fluids, including the contents of chickenpox vesicles, cerebrospinal fluid, and accumulates in milk, amniotic fluid and the placenta. Its concentration in vaginal contents varies widely. Serum concentrations of the drug in mother and newborn are approximately the same. The drug is practically not absorbed through the skin. T1/2 of acyclovir averages 2.5 hours in adults, 4 hours in newborns, and can increase to 20 hours in patients with renal failure. The drug is almost completely excreted unchanged by the kidneys. During pregnancy, the pharmacokinetics of drugs does not change.
As a rule, acyclovir is well tolerated. When using an ointment based on polyethylene glycol, irritation of the genital mucosa and a burning sensation are possible. When taken orally, the drug occasionally causes headache, dizziness, rash and diarrhea. Renal failure and neurotoxic effects are even less common. Side effects of valacyclovir are similar to those of acyclovir - nausea, diarrhea, headache; high doses may cause confusion, hallucinations, kidney damage and, very rarely, thrombocytopenia. With intravenous administration of large doses of acyclovir, renal failure and central nervous system damage may develop.
Famciclovir itself is inactive, but upon its first passage through the liver it is quickly converted to penciclovir. Penciclovir is an acyclic analogue of guanosine. The mechanism of action of the drug is similar to the mechanism of action of acyclovir. Like acyclovir, penciclovir acts primarily against herpes simplex viruses and Varicella-zostervirus. Resistance to penciclavir is rare in the clinic.
Unlike penciclovir, whose bioavailability when taken orally is only 5%, famciclovir is well absorbed. When taking famciclovir, the bioavailability of penciclovir increases to 65–77%. Eating together with the drug slows down the absorption of the latter, but in general the bioavailability is not reduced. The volume of distribution of penciclovir is 2 times the volume of fluid in the body, T1/21/2 increases to 9.9 hours. The drug is easily removed by hemodialysis.
Acyclovir is well tolerated, but sometimes headache, nausea, diarrhea, urticaria may occur, and in older people - hallucinations and confusion. Topical preparations may cause contact dermatitis and ulceration.
The safety of the drug during pregnancy, as well as its interaction with other drugs, has not been established.
Ganciclovir is an acyclic analogue of guanosine. The mechanism of action of the drug is similar to the mechanism of action of acyclovir. Active against all herpes viruses, but most effective against cytomegalovirus.
The bioavailability of ganciclovir when taken orally with food is 6–9% and slightly less when taken on an empty stomach. Valganciclovir is well absorbed and quickly hydrolyzed to ganciclovir, the bioavailability of which increases to 61%. When taking valganciclovir with food, the bioavailability of ganciclovir increases by another 25%. With normal renal function, T1/2 is 2–4 hours. More than 90% of the drug is excreted unchanged by the kidneys. In case of renal failure, T1/2 increases to 28–40 hours.
The main dose-limiting side effect of ganciclovir is inhibition of hematopoiesis (neutropenia, thrombocytopenia). In 5–15% of patients, central nervous system lesions of varying severity are noted (from headache to seizures and coma). With intravenous administration, phlebitis, azotemia, anemia, rashes, fever, changes in liver biochemical parameters, nausea, vomiting, and eosinophilia are possible.
In laboratory animals, the drug had a teratogenic and embryotoxic effect and irreversibly impaired reproductive function. Cytotoxic drugs increase the side effects of ganciclovir on the bone marrow.
Idoxuridine is an iodine-containing analogue of thymidine. The mechanism of antiviral action is not fully understood. It is known that phosphorylated derivatives of the drug are incorporated into viral and cellular DNA, but only inhibit the replication of viral DNA. At the same time, DNA becomes more fragile, easily destroyed, and errors occur more often during its transcription. Resistant strains are isolated from patients with herpetic keratitis treated with idoxuridine. The drug is approved only for topical use. When using it, pain, itching, inflammation and swelling in the eye area, and allergic reactions are possible.
Advances in antimicrobial therapy in the 20th century led to almost complete control of bacterial infections. The task of infectious disease specialists and pharmacologists of the 21st century is to ensure control over viral infection. In addition to being highly effective, new antiviral drugs must be well tolerated. Currently, new agents with fundamentally new mechanisms of action are being developed. Means for suppressing pathological immune reactions and immunotherapy with monoclonal antibodies and vaccines may be promising.
N. M. Kiseleva, Candidate of Medical Sciences, Associate Professor L. G. Kuzmenko, Doctor of Medical Sciences, Professor of Russian State Medical University, Moscow